This site contains some simple Matlab programs that may be useful when computing pseudosections and oxygen fugacity, as well as thermodynamic data files for Theriak-Domino (de Capitani & Petrakakis, 2010). Theriak-Domino is an extremely powerful software package for making pseudosections and performing a range of other thermodynamic calculations.
All codes and input files are free of charge for non-commercial purposes and are to be run at the sole responsibility of the user. Please let me know if you find any bugs.
The Theriak-Domino homepage of Prof. Dr. C.E. de Capitani is here, and a helpful Research Talk site is here.
Codes
apca.m
This code corrects bulk-rock CaO wt% for the amount of CaO contained in apatite. It uses the CaO/P2O5 weight ratio of typical fluorapatite. This can be changed if desired in the code itself.
feloi.m
This code takes bulk-rock Fe2O3 wt% (as usually determined by XRF), and converts it to FeO and Fe2O3 wt%’s given the user’s input molar Fe3+/(Fe2+ + Fe3+) ratio. The loss on ignition (LOI) value is also corrected for the amount of oxygen mass gained during ignition when all the FeO in the rock is converted to Fe2O3.
genmolo2.m
Given input bulk-rock wt% values, this code computes molar compositions suitable for pseudosection input, including formatting that can be used directly with Theriak-Domino. Both the Theriak-Domino and Thermocalc ferrous-ferric styles are output as well. Theriak-Domino formatting for MnO projected out through spessartine is also provided.
A typical sequence of execution would be to run apca and feloi to get the CaO, FeO, Fe2O3, and corrected LOI contents for the rock. Then run genmolo2 to compute the molar composition.
calc_fo2_muo2.m
This code calculates the oxygen fugacity given an input temperature and chemical potential of O2 (mu_O2) at the P-T of interest for the temperature range 150 to 1200 oC. The chemical potential can be obtained using a very nice feature of recent versions of Theriak. Do a calculation using print-code 1 (i.e., with a 1 starting the composition input line, to give detailed output). Near the end of the output, the program will print out the chemical potentials in the system. If there is redox involving ferrous-ferric iron, then O2 will be one of the chemical potentials given. Copy this value into the program calc_fo2_muo2 and input the temperature. The program will printout the ln(fO2) and log10(fO2) values.
Theriak-Domino input files
These files are used in our group. They are based on tcds62cid provided with release 11.03.2020, which uses the dataset of Holland and Powell (2011; the Thermocalc page is here). tcds62cid is in turn based on the input files of Prof. D.K. Tinkham, which can be found here.
The files incorporate a number of updates and various edits. These include updating the garnet parameters for mafic systems (no pressure dependence) and the delG(tran) offset for the margarite endmember in muscovite (5000 J/mol) (see the Thermocalc datafile tc-mb50NCKFMASHTO.txt). Things that have been edited are flagged with “jja” in the files.
The data files also incorporate non-ideal H2O–CO2 mixing according to Holland & Powell (2003). Unless you are doing calculations at high temperatures and very low pressures, this must be accounted for because H2O–CO2 mixtures are highly non-ideal. For example, activity coefficients for CO2 in water-rich subduction zone fluids can be 5–10.
All files have zoisite and microcline deactivated, but these can be re-enabled by removing the commented-out lines (beginning with !). Endmember hematite and ilmenite are also deactivated, in favor of the solid solution models (see below).
To activate a solution phase, change MuNERAL DATA to MINERAL DATA, SuLUTION DATA to SOLUTION DATA, and MuRGULES PARAMETER to MARGULES PARAMETER. To deactivate, do the opposite.
The “mafic” files enable the mafic melt, amphibole, and pyroxene models of Green et al. (2016). There are two pyroxene varieties: CPXo uses omphacitic clinopyroxene, whereas CPXg uses augitic clinopyroxene. The latter incorporates tetrahedral Al and is suitable for high-temperature mafic systems that contain melt. Theriak-Domino 11.03.2020 uses “external” coding in subroutine fsol.f90 to compute the amphibole and augite solutions. The “aug” input files use CPXg, and the “omp” files use CPXo. Solution models for phases relevant to more aluminous metapelitic compositions are deactivated (staurolite, cordierite, chloritoid, and sapphirine).
The “pelitic” files enable the felsic melt model in White et al. (2014), aluminous metapelitic solid solutions, and CPXo for clinopyroxene.
The comparison below shows excellent agreement between Domino using CPXg and Figure 3a (high- and low-pressure parts) of Green et al. (2016). The red (high-P) and orange (low-P) lines are the ones calculated by Domino. Slight differences involve the updated thermodynamic parameters for garnet and white mica, and the consideration of ternary (K-bearing) plagioclase in the Domino calculations. Input file: tcds62cid_mafic_aug_noMn. The bulk compositions used are:
SI(59.76)TI(1.26)AL(6.59)AL(6.59)FE(1.62)FE(4.92)MG(9.88)CA(8.27)NA(1.82)NA(1.82)K(0.44)K(0.44)H(4.62)H(4.62)O(174.19) * SQA (Low-P)
SI(60.05)TI(1.27)AL(6.62)AL(6.62)FE(0.66)FE(5.91)MG(9.93)CA(8.31)NA(1.83)NA(1.83)K(0.44)K(0.44)H(4.64)H(4.64)O(174.55) * SQA (High-P)
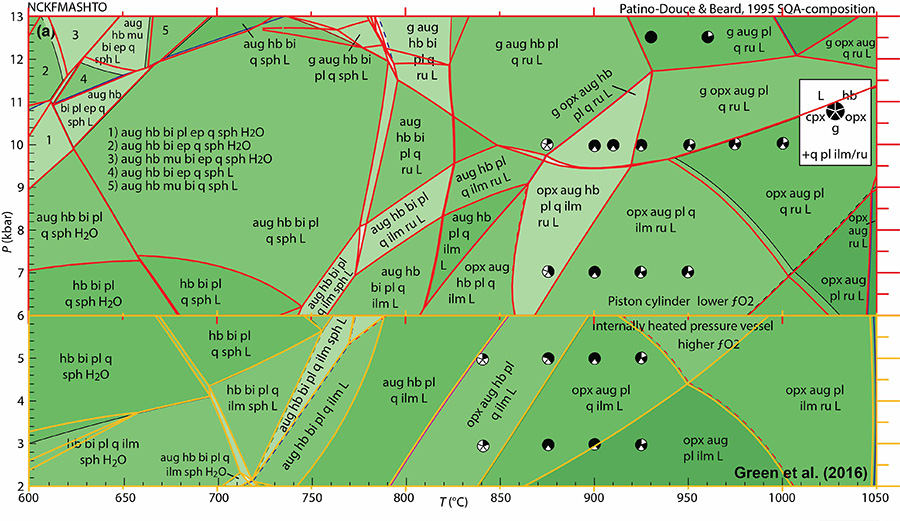
There are three varieties of ilmenite model in these files: ILM, ILM00Mn (default), and ILM00. ILM incorporates Mn and Mg, ILM00Mn just Mn, and ILM00 no Mg or Mn. This is because for some compositions, the activity model appears to incorporate too much Mg, particularly at high temperatures. The ilmenite models seem to run best when SEEDS are provided. An example is given in the files. Because many examples published in the literature (e.g., Green et al., 2016) use ilmenite–hematite mixing with no Mn or Mg, input files that use ILM00 are provided for convenience (“ilm00” filenames).
Versions of the files with Mn-bearing solid solutions deactivated are also provided (“noMn” filenames), and should be used when computing pseudosections in Mn-free systems (the noMn files use ILM00 for ilmenite-hematite solid solutions).
When computing pseudosections in systems with no Fe3+, a very small amount of oxygen can be added to the bulk composition to keep the activity models happy. This approach usually works but adds calculation time and can lead to instabilities if the amount of oxidized iron is tiny. An alternative is to deactivate Fe3+ in all the solution models, but care must be taken to also consider/modify any external solution models that include Fe3+.
Download input datafiles here.
References
de Capitani, C. & Petrakakis, K. (2010). The computation of equilibrium assemblage diagrams with Theriak/Domino software. American Mineralogist 95, 1006-1016.
Green, E.C.R., White, R.W., Diener, J.F.A., Powell, R., Holland, T.J.B. et al. (2016). Activity-composition relations for the calculation of partial melting equilibria in metabasic rocks. Journal of Metamorphic Geology 34, 845-869.
Holland, T.J.B. & Powell, R. (2003). Activity–composition relations for phases in petrological calculations: an asymmetric multicomponent formulation. Contributions to Mineralogy and Petrology 145, 492-501.
Holland, T.J.B. & Powell, R. (2011). An improved and extended internally consistent thermodynamic dataset for phases of petrological interest, involving a new equation of state for solids. Journal of Metamorphic Geology 29, 333-383.
White, R.W., Powell, R., Holland, T.J.B., Johnson, T.E. & Green, E.C.R. (2014). New mineral activity-composition relations for thermodynamic calculations in metapelitic systems. Journal of Metamorphic Geology, 32, 261-286.